Das seltene Gorham-Stout-Syndrom
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Osteolytische Knochenveränderungen finden sich im Wachstumsalter häufig. Es gibt zahlreiche Differenzialdiagnosen, die in der weiteren Abklärung berücksichtigt werden müssen. Eine kurze Übersicht wird in diesem Artikel gegeben. Anhand eines Fallberichts wird auf die seltene Differenzialdiagnose der Gorham-Stout-Erkrankung eingegangen.
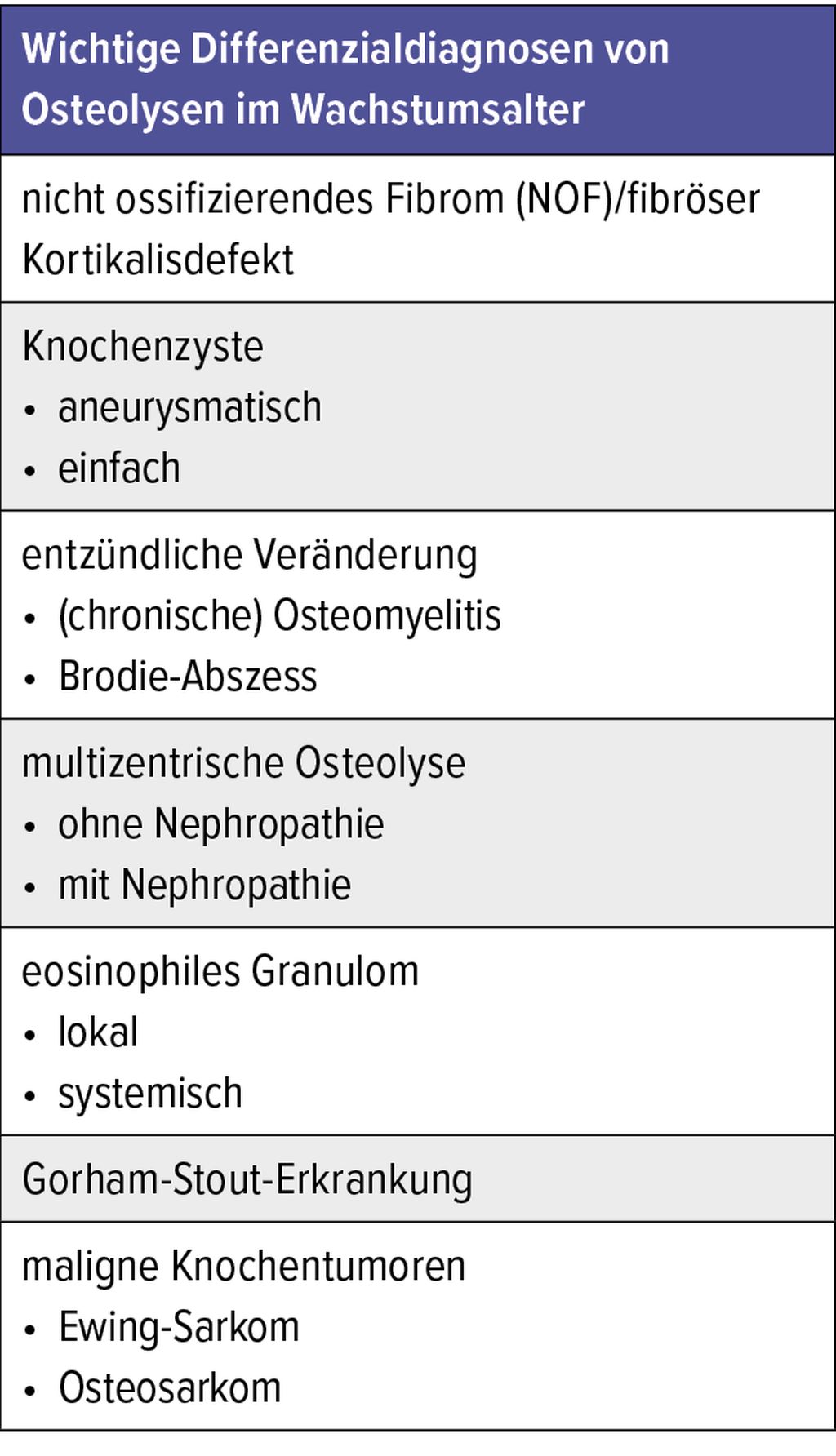
Übersicht über Differenzialdiagnosen bei Osteolysen
Eine Osteolyse ist definiert als Knochenresorption und kann durch tumoröse oder auch nicht tumoröse Veränderungen am Knochen verursacht werden. Im Wachstumsalter finden sich osteolytische Veränderungen nicht so selten. Einige von ihnen sind bedenkenlos, bedürfen nicht einmal der weiteren Abklärung, andere hingegen müssen genauer unter die Lupe genommen werden und haben lokale oder gar systemische Therapiekonsequenzen.
Sehr häufig finden sich im Wachstumsalter ein NOF (nicht ossifizierendes Fibrom) bzw. der fibröse Kortikalisdefekt. Beide stellen meist radiologische Zufallsbefunde dar. Sie haben primär keinen pathologischen Wert und bedürfen als „Leave me alone“-Läsion in der Regel auch keiner Therapie – ja eigentlich nicht einmal einer Kontrolle.
Tab. 1
Aneurysmatische Knochenzysten haben im Wachstumsalter eine Inzidenz von 1,4:1000000 und machen ca. 6% der primären Knochenläsionen aus.1 Die Inzidenz der einfachen Knochenzyste hingegen ist nicht sicher bekannt, da sie oft stumm bleibt. Sie ist häufig ein Zufallsbefund oder fällt im Rahmen einer pathologischen Fraktur auf. Der betroffene Knochen zeigt eine lokale Osteolyse.
Das eosinophile Granulom ist mit einer Inzidenz von 5:1000000 eine seltene Entität2, die ebenfalls zu osteolytischen Veränderungen führt. Es kann lokal oder auch multiostotisch als Langerhanszell-Histiozytose auftreten.
Maligne Knochentumoren machen etwa 3% aller Tumoren im Wachstumsalter aus3 und führen ebenfalls zu Osteolysen. Das Osteosarkom und das Ewingsarkom sind hierbei führend.
Weitaus häufigere Ursache für Osteolysen im Wachstumsalter sind entzündliche Veränderungen, die vor allem in ihrer chronischen Form entweder lokal begrenzt (Brodie-Abszess) oder multipel (chronische Osteomyelitis) auftreten.
Osteolysen fallen entweder als Zufallsbefunde im Rahmen einer Schmerzabklärung auf oder auch dann, wenn der Knochen bereits so geschwächt ist, dass er bricht und sich eine pathologische Fraktur ereignet.
Fallbericht
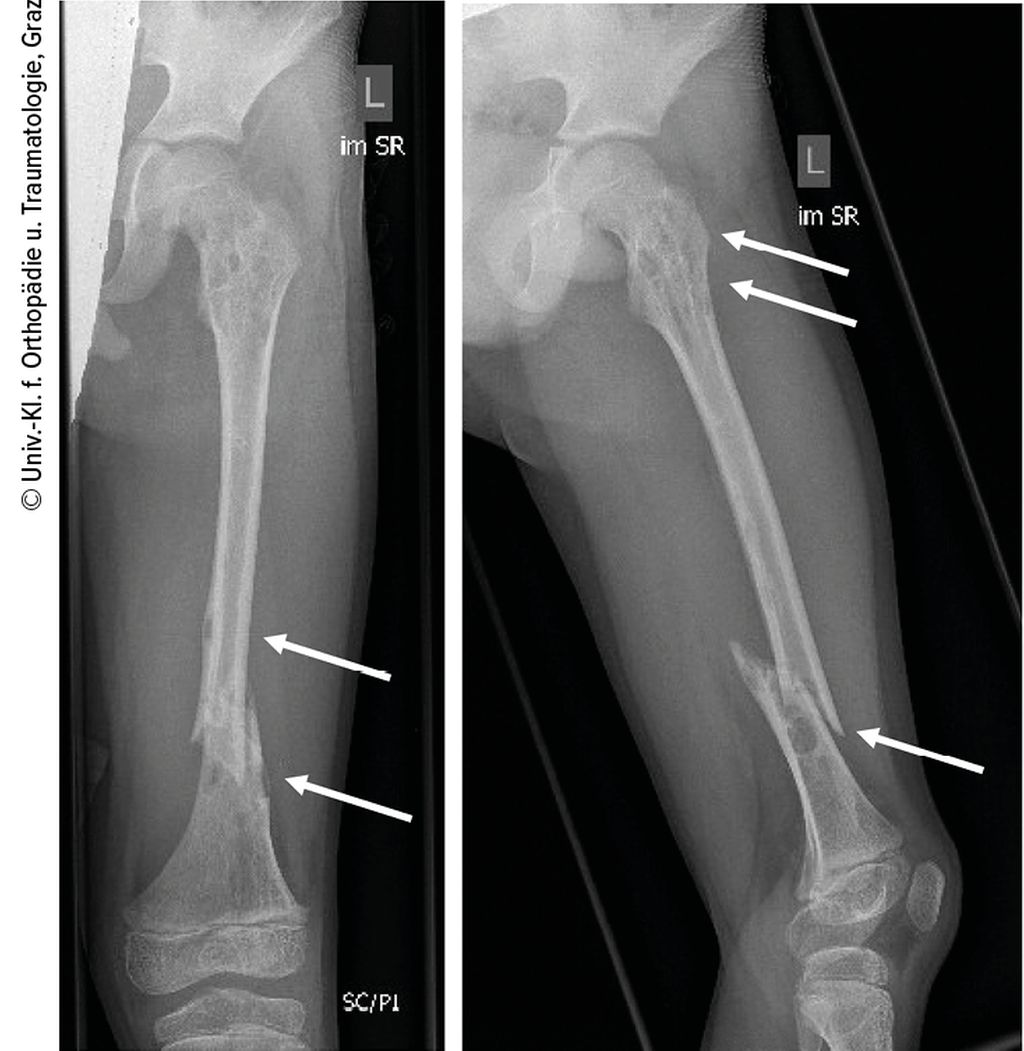
Abb. 1: Distale Femurschaftschrägfraktur links und zahlreiche Osteolysen im Röntgen
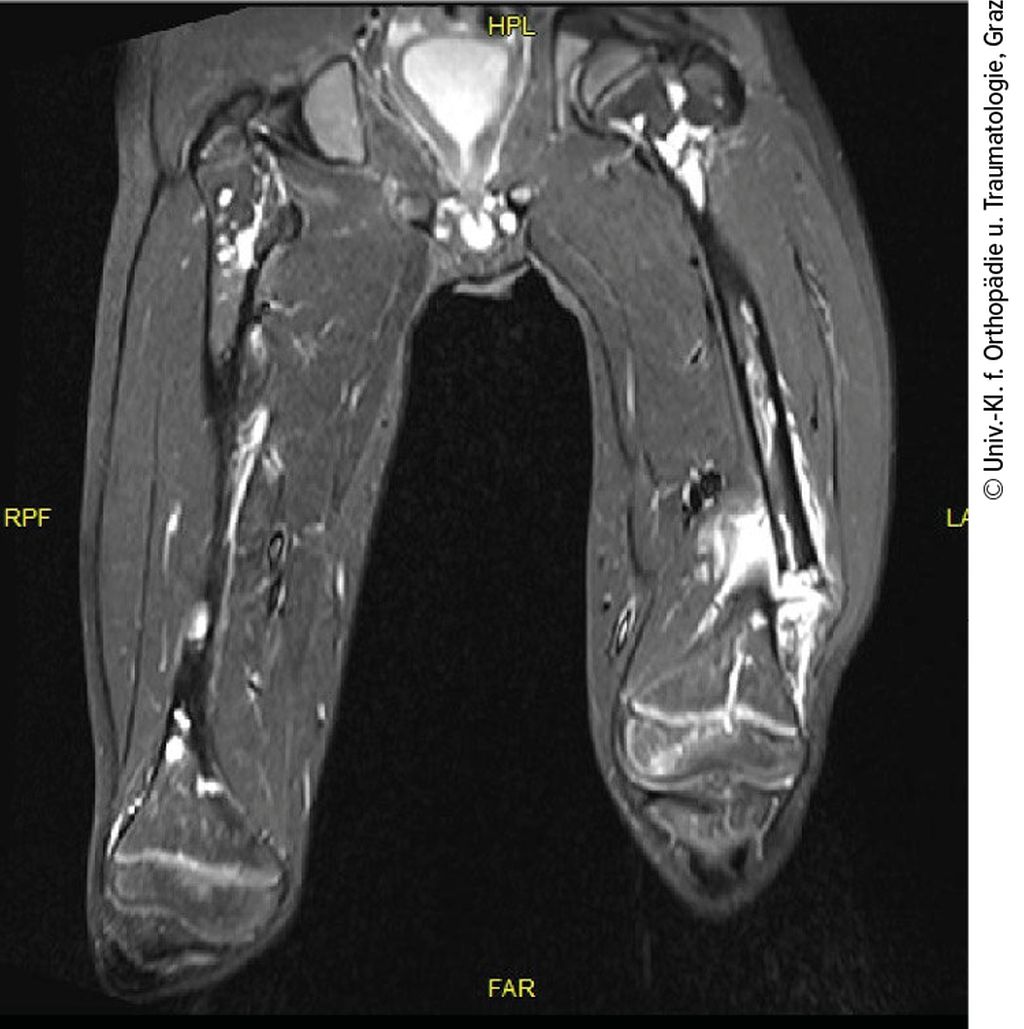
Abb. 2: MR-tomografische Darstellung der osteolytischen Läsionen an beiden Oberschenkeln
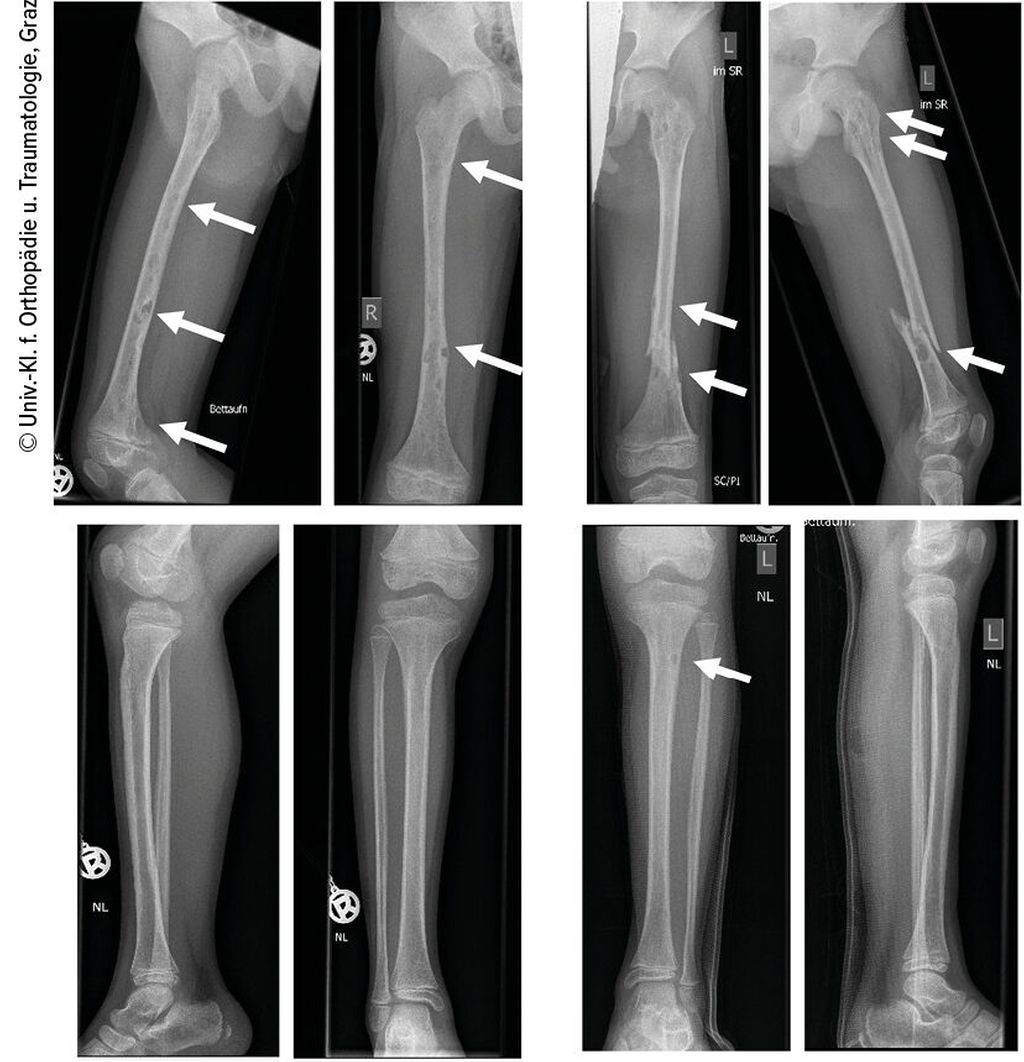
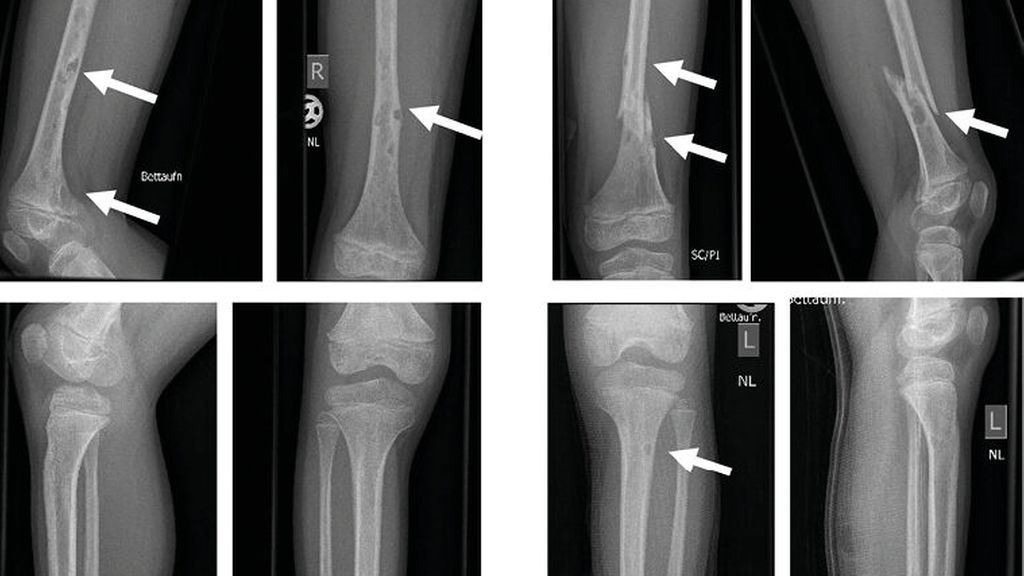
Abb. 3: Röntgenbilder beider Unterschenkel und des rechten Oberschenkels
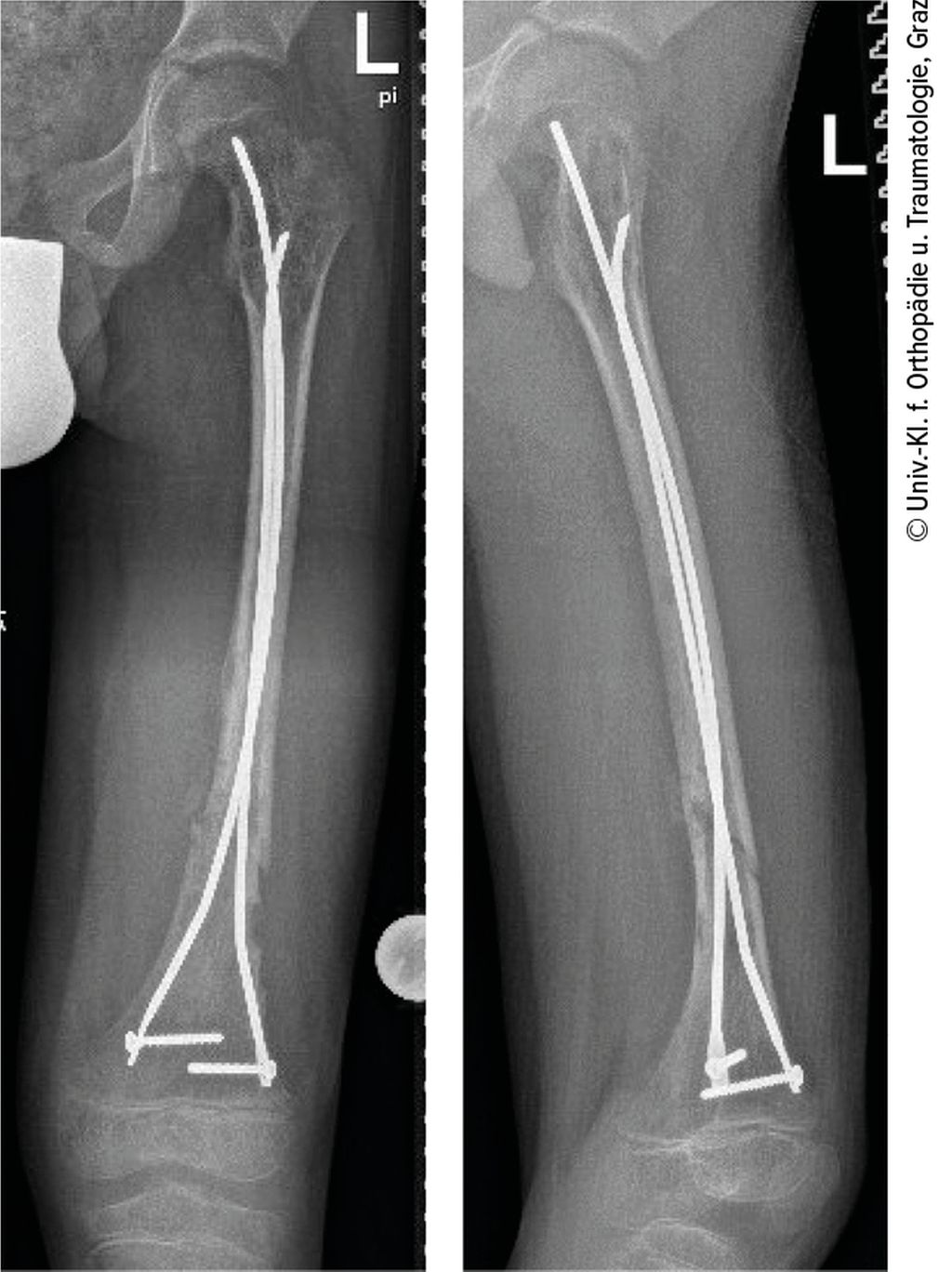
Abb. 4: Stabilisierung der Fraktur mittels zweier verriegelter ESINs
Ein 10-jähriger Bub wurde nach Kollision beim Bobfahren per Rettung in die Ambulanz gebracht. Die Schwellung, Verkürzung und Fehlstellung des linken Femurs ließen eine Femurfraktur vermuten. Nach Durchführung des Röntgens bestätigte sich eine distale Femurschaftschrägfraktur, allerdings wies der gesamte Knochen auch zahlreiche Osteolysen auf (Abb. 1). Ergänzend erfolgten ein CT und ein MRT (Abb. 2). Die Ursache der pathologischen Oberschenkelfraktur legte den Verdacht einer Gorham-Stout-Erkrankung nahe – ein maligner Prozess konnte allenfalls ausgeschlossen werden. Röntgenbilder beider Unterschenkel und auch des rechten Oberschenkels wurden ergänzend angefertigt (Abb. 3). Auch hier zeigten sich multiple osteolytische Herde, die den Knochen jedoch nicht zu gefährden schienen.
Der Probegewebsentnahme folgte die Stabilisierung der Fraktur mittels zweier verriegelter ESINs („elastic stable intramedullary nails“) (Abb. 4). Der Patient wurde problemlos zunächst unter Teilbelastung über 4 Wochen mobilisiert, die Vollbelastung wurde nach 6 Wochen erreicht. Der postoperative Verlauf zeigte eine gute Kallusbildung und adäquate Knochenheilung.
Die Mutter des Patienten berichtete aus der Vorgeschichte des Buben über unklare zystische Veränderungen am Unterkiefer, die vor einigen Jahren aufgetreten waren und zahnmedizinisch–kieferchirurgisch abgeklärt wurden. Die Verdachtsdiagnose eines Cherubismus bestätigte sich nicht. In Zusammenschau mit dem aktuellen Skelettstatus des Buben passten die zystischen Veränderungen am Kiefer gut zu einer Gorham-Stout-Erkrankung. Das histopathologische Ergebnis der Gewebsprobe bestätigte die radiologische Verdachtsdiagnose, auch wenn die multiostotischen zystischen Veränderungen als Besonderheit zu sehen sind, da die Gorham-Stout-Erkrankung meistens als monostotisch beschrieben ist. In einer interdisziplinären Besprechung im Tumorboard wurde jedoch aufgrund des radiologischen Bildes in Zusammenschau mit der Histologie die Diagnose der Gorham-Stout-Erkrankung favorisiert.
Gorham-Stout-Syndrom
Die Gorham-Stout-Erkrankung (GSD = Gorham-Stout Disease, Gorham-Stout-Syndrom, „vanishing bone disease“, „phantom bone disease“, „Gorham’s disease“), ist eine seltene, nicht erbliche Knochenerkrankung, die durch fortschreitende Osteolyse und Lymphangiomatose gekennzeichnet ist. Radiologisch sind die Osteolysen sowohl im Markraum als auch subkortikal und kortikal zu finden. Im Verlauf können die Osteolysen zu einem vollständigen Verschwinden des Knochens führen.
2018 wurde die GSD in die Liste der Gefäßmalformationen als einfache Form einer Gefäßmalformation aufgenommen (International Society for the Study of Vascular Anomalies, ISVVA). Diese Liste umfasst Blut- und Lymphgefäßveränderungen, wobei die meisten Veränderungen durch somatische PIK3CA-Mutationen verursacht werden. Für die GSD trifft dies allerdings nicht zu. In der Literatur sind bisher etwa 300 GSD-Fälle beschrieben (Orphanet Report Series 2022).
Die GSD gehört zu den idiopathischen Osteolysen und repräsentiert in der Einteilung nach Hardegger einen eigenen Typen (Tab.2).4 Die Erstbeschreibung der GSD erfolgte durch den amerikanischen Arzt L. S. Jackson. Er berichtete im Boston Medical and Surgical Journal (heute: New England Journal of Medicine) über einen 12-jährigen Patienten mit einem „knochenlosen“ Arm.5,6 Der gesamte Humerusknochen war dabei offensichtlich verschwunden, dennoch nutzte der Patient den Arm mit einer beeindruckenden Funktionalität. Röntgenbilder existierten zu diesem Zeitpunkt nicht. Das damals als „rätselhaft“ beschriebene Knochenverschwinden war die Erstbeschreibung der Gorham-Stout-Erkrankung.
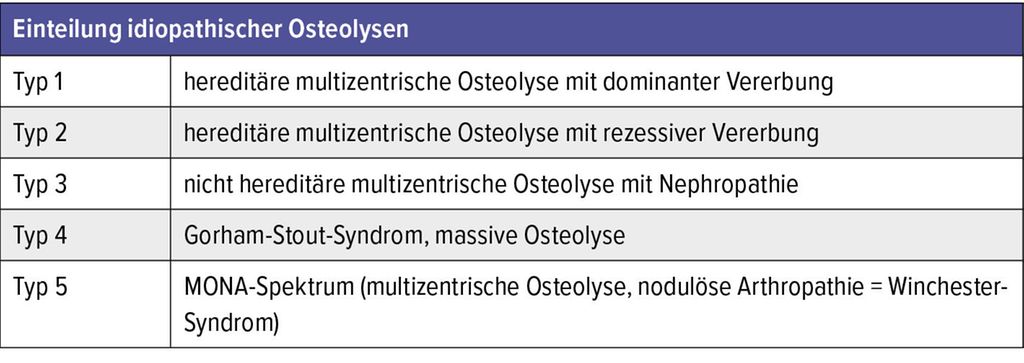
Tab. 2: Einteilung idiopathischer Osteolysen (nach Hardegger et al. 1985)4
Lemuel Whittington Gorham und Arthur Purdy Stout, beide angesehene Ärzte mit lebenslangem Interesse an der Pathologie und letztendlich die Namensgeber der GSD, verfolgten die Hypothese, dass eine massive Angiomatose den Knochen ersetzte. 1955 beschrieben und definierten sie die klinisch-pathologischen Merkmale und beschrieben sie bei bei 24 Patienten.7 Dabei betrachteten sie die ausgeprägte Proliferation dünnwandiger Kapillaren in den Läsionen (die sie „Hämangiomatose“ nannten) als das auffälligste histologische Merkmal der Krankheit. Diese ausgeprägte, unkontrollierte Proliferation von Gefäßendothelzellen ersetzt dabei normales Knochengewebe. Der Krankheitsbeginn erfolgt spontan. Die Erkrankung dauert Jahre an.
Eine Gorham-Stout-Erkrankung tritt häufig bereits im jungen Lebensalter auf, ohne Präferenz für ethnische Merkmale oder Geschlecht.8 Es kann zu lokalen lytischen Ereignissen kommen, es kann aber auch das gesamte Skelett betroffen sein. Die betroffene skelettale Region kann Schmerzen, eine Schwellung oder sogar eine Deformität zeigen. Die Funktion ist in der Regel unbeeinträchtigt. Generalisierte Symptome bestehen nicht. Die GSD kann überall am Skelett gefunden werden, oftmals sind jedoch Schulter- und Beckengürtel sowie die Schädelknochen betroffen. Eine Mitbeteiligung der Wirbelsäule ist möglich, aber eher ungewöhnlich.9
Die Diagnose ist vor allem eine Ausschlussdiagnose und basiert neben der klinischen Untersuchung auf einer Bildgebung. Letztendlich muss sie histopathologisch verifiziert werden. Als Differenzialdiagnose kommen metabolische, endokrine, infektiöse, maligne und immunologische Erkrankungen in Betracht. Auch muss die idiopathische Osteolyse, die an einen spezifischen Genlokus gekoppelt ist und mit oder ohne Nephropathie auftreten kann, ausgeschlossen werden.10 Histologisch zeigt der von der Gorham-Stout-Erkrankung betroffene Knochen eine benigne vaskuläre Proliferation von dünnwandigen lymphatischen und vaskulären Kapillaren und Ausdünnung der Knochentrabekel8 mit konsekutiv sichtbarer Osteolyse sowie fettiger Degeneration des Knochenmarks mit Präsenz spindelförmiger Fibroblasten.7,11
Mehrere Hypothesen wurden für die Ätiopathogenese der GSD aufgestellt: lokale Hypoxie/Azidose, endotheliale Dysplasie, erhöhte osteoklastische Aktivität oder gesteigerte Empfindlichkeit von Osteoklastenvorläufern auf humorale Faktoren, Mangel an Schilddrüsen-C-Zellen und Calcitonin sowie Lymphgefäßproliferation.12
In ihrer 2020 publizierten Arbeit beobachteten Rossi et al., dass 75% der Osteoklasten von GSD-Patient:innen mehr Motilität zeigten, einen veränderten Aufbau aufwiesen (Lamellipodien, Stressfasern und Membrankräuselung) sowie um das 4,5-Fache aktiver waren.13 Auch veränderte Signalwege, die Differenzierung und Funktion von Osteoklasten betreffend, konnten nachgewiesen werden: Angiotensin-II-stimulierte Signalübertragung durch G-Proteine und Beta-Arrestin, PI3-Kinase-Weg und EGF-Rezeptor-Signalweg.13
Auf der Ebene der Osteoblasten kommt es bei Patienten mit GSD zu einer Überexpression von MMP13 (Matrix-Metallopeptidase 13; das Genprodukt ist an der Kapillarbildung und dem osteozytären perilakunären Umbau beteiligt) und RXFP1 (Relaxin-Familie-Peptidrezeptor 1; führt in fibrokartilaginären Zellen zu einem Anstieg von MMP13 und MMP9).13
Die Osteolyse ist charakterisiert als spontan, progressiv und ohne Knochenneubildung. Die histopathologischen Veränderungen zeigen einen deutlichen Verlust der Knochenmatrix mit Proliferation der dünnwandigen kapillär-vaskulären Kanäle oder Proliferation von Bindegewebe.12,14 Nicht immer folgt der signifikante Knochenverlust einer signifikanten vaskulären Proliferation.15,16 Die vaskuläre Proliferation kann auch in das umliegende Gewebe erfolgen.17 Der natürliche Verlauf der Knochenläsionen ist schwer vorhersagbar. Die Knochenresorption kann spontan stoppen – dies passiert am häufigsten. Es gibt Fälle, in denen die Läsionen über Jahrzehnte ohne Reossifikation stabil bleiben.14,18
Behandlungsoptionen
Aufgrund der Seltenheit der Erkrankung gibt es keine standardisierten Behandlungsprotokolle. Es gibt aber Behandlungsversuche pharmakologischer und operativer Art sowie auch die Möglichkeit der Strahlentherapie oder eine Kombination davon.19
Die chirurgischen Verfahren (Knochentransplantationen und -rekonstruktionen, Amputation, Endoprothetik) sind mit allgemeinen Operationskomplikationen verbunden (Infektionen, Nerven- und Gefäßschäden), aber auch mit dem Wiederauftreten von Osteolysen und mit Transplantatresorption.20
Die Pharmakotherapie bestreiten Interferon, Bisphosphonate, Kalziumsalze und Vitamin D, Interferon und Zoledronsäure, Cyclophosphamid und Fluorouracil sowie Calcitonin, Alendronat-Natrium und Sirolimus.19, 21 Sirolimus (Rapamycin) ist ein mTOR-Inhibitor, der eine antiangiogene Aktivität und eine verbessernde Wirkung auf klinische Symptome und die Lebensqualität bei GSD-Patienten hat.22
Eine Strahlentherapie kann das Fortschreiten der Erkrankung offenbar wirksam verhindern.23
Postoperativ wurde bei unserem Patienten – im neuerlichen interdisziplinären Beschluss – eine systemische Therapie mit dem Immunsuppressivum Rapamycin eingeleitet. Hierbei ist die Dosierung so zu wählen, dass das Ausmaß möglicher Nebenwirkungen (z.B. Aphthen, permanente Infekte) gering gehalten wird. Im Falle unseres Patienten zeigte die eingeleitete immunsuppressive Therapie zwar eine Besserung („Verdichtung“) der im Nativröntgenbild sichtbaren Läsionen, aber keine signifikante Reduktion. Bereits mehrere Monate nach der pathologischen Fraktur des distalen linken Oberschenkels beklagte der Patient auch belastungsabhängige Schmerzen im rechten Oberschenkel. Eine prophylaktische Stabilisierung wurde von kinderorthopädischer Seite empfohlen. Diese Maßnahme stellt eine weitere Option dar, um den durch die Osteolysen geschwächten Knochen zu stabilisieren und so dem Patienten einen unbeschwerten Alltag zu ermöglichen. Im Rahmen der Stabilisierungsoperation ist die erneute Probegewebsentnahme geplant.
Eine Strahlentherapie ist bei offenen Fugen allerdings nicht die Therapie der ersten Wahl. Die chirurgische Behandlung mit oder ohne Strahlentherapie wird vor allem bei ausgedehnten Läsionen bevorzugt, insbesondere bei funktioneller Instabilität.24
Perspektiven
In den meisten Fällen schreitet die Krankheit langsam voran, die Prognose bleibt jedoch unvorhersehbar.25 Die alleinige Beteiligung der Gliedmaßen oder des Beckens hat keinen Einfluss auf die Lebenserwartung.18,26 Die Beteiligung bestimmter Knochen, wie Rippen, Schulterblatt und Brustwirbel, kann zu einer Nervenwurzelkompression mit eventuellem Querschnitt führen und lebensbedrohliche Komplikationen hervorrufen.14,27,28 Die Gesamtmortalität wurde in Studien auf 13% geschätzt.25 Bei Mitbeteiligung der Wirbelsäule steigt die Komplikationsrate durch eine mögliche Kompression des Rückenmarks oder aufgrund einer Wirbelinstabilität als Folge einer Osteolyse.14,29 Auch das Vorliegen eines Chylothorax scheint prognostisch ungünstig zu sein.27,30
Schlussfolgerung
Die Gorham-Stout-Erkrankung ist eine multifaktorielle Erkrankung. Sie sollte nach Ausschluss anderer Pathologien mit massiver Osteolyse in Betracht gezogen und histopathologisch bestätigt werden. Häufige Läsionsorte sind Becken- und Schultergürtel sowie die Schädelkalotte.
Osteolytische Läsionen der Wirbelsäule sind selten, bergen aber das Risiko einer möglichen Instabilität und damit Kompression von Spinalnerven. Das Vorhandensein eines Chylothorax ist ein ungünstiges prognostisches Zeichen.
Über die Therapie gibt es derzeit keinen Konsens. Weite operative Resektionen sollten aber vermieden werden. Die pharmakologische Therapie ist bei systemischem Befall mit einzubeziehen.
Literatur:
1 Leithner A et al.: Aneurysmal bone cyst. Clin Orthop Relat Res 1999; (363): 176-9 2 Zehetgruber H et al.: Prevalence of aneurysmal and solitary bone cysts in young patients. Clin Orthop Relat Res 2005; 439: 136-43 3 Jackson TM et al.: Pediatric malignant bone tumors. Curr Probl Pediatr Adolesc Health Care 2016; 46(7): 213-28 4 Hardegger F et al.: The syndrome of idiopathic osteolysis. J Bone Joint Surg Br 1985; 67(1): 89-93 5 Jackson JBS: A boneless arm. New Engl J Med 1838; 368-9 6 Jackson JBS: Absorption of the humerus after fracture. New Eng J Med 1872; 87: 245-7 7 Gorham LW, Stout AP: Massive osteolysis (acute spontaneous absorption of bone, phantom bone, disappearing bone); its relation to hemangiomatosis. J Bone Joint Surg Am 1955; 37-A(5): 985-1004 8 Ruggieri P et al.: Gorham-Stout disease: the experience of the Rizzoli Institute and review of the literature. Skeletal Radiol 2011; 40(11): 1391-7 9 Flörchinger A et al.: Das Gorham-Stout-Syndrom der Wirbelsäule. Rofo 1998; 168(1): 68-76 10 Tsang WM et al.: Massive osteolysis (Gorham disease) of the maxillofacial skeleton. J Oral Maxillofac Surg 2004; 62(2): 225-30 11 Möller G et al.: The Gorham-Stout syndrome (Gorham’s massive osteolysis). J Bone Joint Surg Br 1999; 81(3): 501-6 12 Chrcanovic BR, Gomez RS: Gorham-Stout disease with involvement of the jaws. Int J Oral Maxillofac Surg 2019; 48(8): 1015-21 13 Rossi M et al.: Dissecting the mechanisms of bone loss in Gorham-Stout disease. Bone 2020; 130: 115068 14 Boyer P et al.: Massive Gorham-Stout syndrome of the pelvis. Clin Rheumatol 2005; 24(5): 551-5 15 Kamble P et al.: A rare case of vanishing bone disease of metacarpals. J Orthop Case Rep 2021; 11(1): 101-3 16 Kawasaki K et al.: Is angiomatosis an intrinsic pathohistological feature of massive osteolysis? Virchows Arch 2003; 442(4): 400-6 17 Gondivkar SM, Gadbail AR: Gorham-Stout syndrome: arare clinical entity and review of literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2010; 109(2): e41-8 18 Rauh G, Gross M: Disappearing bone disease (Gorham-stout disease). Eur J Med Res 1997; 2(10): 425-7 19 Ellati R et al.: Novel approach of treating gorham-stout disease in the humerus. Eur Rev Med Pharmacol Sci 2016; 20(3): 426-32 20 Woodward HR et al.: Massive osteolysis of the cervical spine. Spine 1981; 6(6): 545-9 21 Rossi M et al.: Dysregulated miRNAs in bone cells of patients with Gorham-Stout disease. FASEB J 2021; 35(3): e21424 22 Ozeki M et al.: The impact of sirolimus therapy on lesion size, clinical symptoms, and quality of life of patients with lymphatic anomalies. Orphanet J Rare Dis 2019; 14(1): 141 23 Heyd R et al.: Radiation therapy for Gorham-Stout syndrome: results of a national patterns-of-care study and literature review. Int J Radiat Oncol Biol Phys 2011; 81(3): e179-85 24 Nikolaou VS et al.: Vanishing bone disease (Gorham-Stout syndrome). World J Orthop 2014; 5(5): 694-8 25 Bode-Lesniewska B et al.: Gorham-Stout disease of the shoulder girdle and cervico-thoracic spine: fatal course in a 65-year-old woman. Skeletal Radiol 2002; 31(12): 724-9 26 Turra S et al.: A 20-year follow-up study of a case of surgically treated massive osteolysis. Clin Orthop Relat Res 1990; 250: 297-302 27 Liu Y et al.: Gorham-Stout disease: radiological, histological, and clinical features of 12 cases and review of literature. Clin Rheumatol 2016; 35(3): 813-23 28 Liu M et al.: Mandibular Gorham-Stout disease. Medicine 2017; 96(42): e8184 29 Lee S et al.: Gorham Stout syndrome (disappearing bone disease): two additional case reports and a review of the literature. Arch Otolaryngol Head Neck Surg 2003; 129(12): 1340-3 30 Shimizu T et al.: A case report of Gorham-Stout syndrome remission. J Orthop Sci 2012; 17(2): 199-204
Das könnte Sie auch interessieren:
Die Pavlik-Riemenbandage
Seit dem Jahr 1991 ist in Österreich eine orientierende Hüftsonografie des Neugeborenen in der 1. sowie in der 6.–8. Lebenswoche ein fester Bestandteil der routinemäßigen Untersuchungen ...
Therapie von Fragilitätsfrakturen des Beckenringes
FFP-Frakturen („fragility fractures of the pelvis“) entsprechen in Morphologie und Entstehungsmechanismus den osteoporotischen Beckenbrüchen – so wurden diese Frakturen über Jahrzehnte ...

Das stabile Handgelenk – Übungsprogramme und deren Effekte
Ein instabiles Handgelenk resultiert aus einer Dysfunktion des Zusammenspiels der entsprechenden Strukturen. Durch die Anwendung von darauf abgestimmten Übungsprogrammen kann das ...